Storage for one atomic configuration. More...
#include <Structure.h>
Public Types | |
| enum | SampleType { ST_UNKNOWN , ST_TRAINING , ST_VALIDATION , ST_TEST } |
| Enumerates different sample types (e.g. More... | |
Public Member Functions | |
| Structure () | |
| Constructor, initializes to zero. | |
| void | setElementMap (ElementMap const &elementMap) |
| Set element map of structure. | |
| void | addAtom (Atom const &atom, std::string const &element) |
| Add a single atom to structure. | |
| void | readFromFile (std::string const fileName="input.data") |
| Read configuration from file. | |
| void | readFromFile (std::ifstream &file) |
| Read configuration from file. | |
| void | readFromLines (std::vector< std::string > const &lines) |
| Read configuration from lines. | |
| void | calculateMaxCutoffRadiusOverall (EwaldSetup &ewaldSetup, double rcutScreen, double maxCutoffRadius) |
| Calculate maximal cut-off if cut-off of screening and real part Ewald summation are also considered. | |
| void | calculateNeighborList (double cutoffRadius, bool sortByDistance=false) |
| Calculate neighbor list for all atoms. | |
| void | calculateNeighborList (double cutoffRadius, std::vector< std::vector< double > > &cutoffs) |
| Calculate neighbor list for all atoms and setup neighbor cut-off map. | |
| void | sortNeighborList () |
| Sort all neighbor lists of this structure with respect to the distance. | |
| void | setupNeighborCutoffMap (std::vector< std::vector< double > > cutoffs) |
| Set up a neighbor cut-off map which gives the index (value) of the last needed neighbor corresponding to a specific cut-off (key). | |
| void | calculatePbcCopies (double cutoffRadius, int(&pbc)[3]) |
| Calculate required PBC copies. | |
| void | calculateInverseBox () |
| Calculate inverse box. | |
| bool | canMinimumImageConventionBeApplied (double cutoffRadius) |
| Check if cut-off radius is small enough to apply minimum image convention. | |
| Vec3D | applyMinimumImageConvention (Vec3D const &dr) |
| Calculate distance between two atoms in the minimum image convention. | |
| void | calculateVolume () |
| Calculate volume from box vectors. | |
| double | calculateElectrostaticEnergy (EwaldSetup &ewaldSetup, Eigen::VectorXd hardness, Eigen::MatrixXd gammaSqrt2, Eigen::VectorXd sigmaSqrtPi, ScreeningFunction const &fs, double const fourPiEps, ErfcBuf &erfcBuf) |
| Compute electrostatic energy with global charge equilibration. | |
| double | calculateScreeningEnergy (Eigen::MatrixXd gammaSqrt2, Eigen::VectorXd sigmaSqrtPi, ScreeningFunction const &fs, double const fourPiEps) |
| Calculate screening energy which needs to be added (!) to the electrostatic energy in order to remove contributions in the short range domain. | |
| void | calculateDAdrQ (EwaldSetup &ewaldSetup, Eigen::MatrixXd gammaSqrt2, double const fourPiEps, ErfcBuf &erfcBuf) |
| Calculates derivative of A-matrix with respect to the atoms positions and contract it with Q. | |
| void | calculateDQdChi (std::vector< Eigen::VectorXd > &dQdChi) |
| Calculates derivative of the charges with respect to electronegativities. | |
| void | calculateDQdJ (std::vector< Eigen::VectorXd > &dQdJ) |
| Calculates derivative of the charges with respect to atomic hardness. | |
| void | calculateDQdr (std::vector< size_t > const &atomsIndices, std::vector< size_t > const &compIndices, double const maxCutoffRadius, std::vector< Element > const &elements) |
| Calculates derivative of the charges with respect to the atom's position. | |
| void | calculateElectrostaticEnergyDerivatives (Eigen::VectorXd hardness, Eigen::MatrixXd gammaSqrt2, Eigen::VectorXd sigmaSqrtPi, ScreeningFunction const &fs, double const fourPiEps) |
| Calculates partial derivatives of electrostatic Energy with respect to atom's coordinates and charges. | |
| Eigen::VectorXd const | calculateForceLambdaTotal () const |
| Calculate lambda_total vector which is needed for the total force calculation in 4G NN. | |
| Eigen::VectorXd const | calculateForceLambdaElec () const |
| Calculate lambda_elec vector which is needed for the electrostatic force calculation in 4G NN. | |
| void | remap () |
| Translate all atoms back into box if outside. | |
| void | remap (Atom &atom) |
| Translate atom back into box if outside. | |
| void | toNormalizedUnits (double meanEnergy, double convEnergy, double convLength, double convCharge) |
| Normalize structure, shift energy and change energy, length and charge unit. | |
| void | toPhysicalUnits (double meanEnergy, double convEnergy, double convLength, double convCharge) |
| Switch to physical units, shift energy and change energy, length and charge unit. | |
| std::size_t | getMaxNumNeighbors () const |
| Find maximum number of neighbors. | |
| void | freeAtoms (bool all, double const maxCutoffRadius=0.0) |
| Free symmetry function memory for all atoms, see free() in Atom class. | |
| void | reset () |
| Reset everything but elementMap. | |
| void | clearNeighborList () |
| Clear neighbor list of all atoms. | |
| void | clearElectrostatics (bool clearDQdr=false) |
| Clear A-matrix, dAdrQ and optionally dQdr. | |
| void | updateError (std::string const &property, std::map< std::string, double > &error, std::size_t &count) const |
| Update property error metrics with this structure. | |
| std::string | getEnergyLine () const |
| Get reference and NN energy. | |
| std::vector< std::string > | getForcesLines () const |
| Get reference and NN forces for all atoms. | |
| std::vector< std::string > | getChargesLines () const |
| Get reference and NN charges for all atoms. | |
| void | writeToFile (std::string const fileName="output.data", bool const ref=true, bool const append=false) const |
| Write configuration to file. | |
| void | writeToFile (std::ofstream *const &file, bool const ref=true) const |
| Write configuration to file. | |
| void | writeToFileXyz (std::ofstream *const &file) const |
| Write configuration to xyz file. | |
| void | writeToFilePoscar (std::ofstream *const &file) const |
| Write configuration to POSCAR file. | |
| void | writeToFilePoscar (std::ofstream *const &file, std::string const elements) const |
| Write configuration to POSCAR file. | |
| std::vector< std::string > | info () const |
| Get structure information as a vector of strings. | |
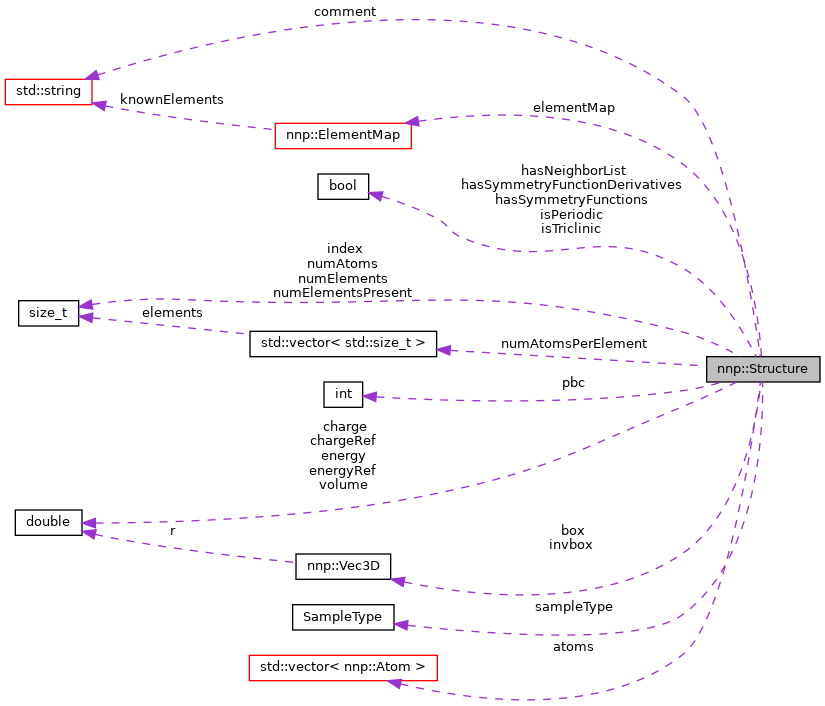
Public Attributes | |
| ElementMap | elementMap |
| Copy of element map provided as constructor argument. | |
| bool | isPeriodic |
| If periodic boundary conditions apply. | |
| bool | isTriclinic |
| If the simulation box is triclinic. | |
| bool | hasNeighborList |
| If the neighbor list has been calculated. | |
| bool | NeighborListIsSorted |
| If the neighbor list has been sorted by distance. | |
| bool | hasSymmetryFunctions |
| If symmetry function values are saved for each atom. | |
| bool | hasSymmetryFunctionDerivatives |
| If symmetry function derivatives are saved for each atom. | |
| std::size_t | index |
| Index number of this structure. | |
| std::size_t | numAtoms |
| Total number of atoms present in this structure. | |
| std::size_t | numElements |
| Global number of elements (all structures). | |
| std::size_t | numElementsPresent |
| Number of elements present in this structure. | |
| int | pbc [3] |
| Number of PBC images necessary in each direction for max cut-off. | |
| double | energy |
| Potential energy determined by neural network. | |
| double | energyRef |
| Reference potential energy. | |
| double | energyShort |
| Short-range part of the potential energy predicted by NNP. | |
| double | energyElec |
| Electrostatics part of the potential energy predicted by NNP. | |
| bool | hasCharges |
| If all charges of this structure have been calculated (and stay the same, e.g. | |
| double | charge |
| Charge determined by neural network potential. | |
| double | chargeRef |
| Reference charge. | |
| double | volume |
| Simulation box volume. | |
| double | maxCutoffRadiusOverall |
| Maximum cut-off radius with respect to symmetry functions, screening function and Ewald summation. | |
| double | lambda |
| Lagrange multiplier used for charge equilibration. | |
| SampleType | sampleType |
| Sample type (training or test set). | |
| std::string | comment |
| Structure comment. | |
| Vec3D | box [3] |
| Simulation box vectors. | |
| Vec3D | invbox [3] |
| Inverse simulation box vectors. | |
| Eigen::MatrixXd | A |
| Global charge equilibration matrix A'. | |
| bool | hasAMatrix |
| If A matrix of this structure is currently stored. | |
| std::vector< std::size_t > | numAtomsPerElement |
| Number of atoms of each element in this structure. | |
| std::vector< Atom > | atoms |
| Vector of all atoms in this structure. | |
Detailed Description
Storage for one atomic configuration.
Definition at line 38 of file Structure.h.
Member Enumeration Documentation
◆ SampleType
Enumerates different sample types (e.g.
training or test set).
| Enumerator | |
|---|---|
| ST_UNKNOWN | Sample type not assigned yet. |
| ST_TRAINING | Structure is part of the training set. |
| ST_VALIDATION | Structure is part of validation set (currently unused). |
| ST_TEST | Structure is part of the test set. |
Definition at line 42 of file Structure.h.
Constructor & Destructor Documentation
◆ Structure()
| Structure::Structure | ( | ) |
Constructor, initializes to zero.
Definition at line 35 of file Structure.cpp.
References box, charge, chargeRef, comment, energy, energyElec, energyRef, energyShort, hasAMatrix, hasCharges, hasNeighborList, hasSymmetryFunctionDerivatives, hasSymmetryFunctions, index, invbox, isPeriodic, isTriclinic, maxCutoffRadiusOverall, NeighborListIsSorted, numAtoms, numElements, numElementsPresent, pbc, sampleType, ST_UNKNOWN, and volume.
Member Function Documentation
◆ setElementMap()
| void Structure::setElementMap | ( | ElementMap const & | elementMap | ) |
Set element map of structure.
- Parameters
-
[in] elementMap Reference to a map containing all possible (symbol, index)-pairs (see ElementMap).
Definition at line 71 of file Structure.cpp.
References elementMap, numAtomsPerElement, and numElements.
Referenced by nnp::Dataset::distributeStructures(), main(), and nnp::Dataset::prepareNumericForces().
◆ addAtom()
| void Structure::addAtom | ( | Atom const & | atom, |
| std::string const & | element ) |
Add a single atom to structure.
- Note
- Be sure to set the element map properly before adding atoms. This function will only keep the atom's coordinates, energy, charge, tag and forces, all other members will be cleared or reset (in particular, the neighbor list and symmetry function data will be deleted).
Definition at line 80 of file Structure.cpp.
References atoms, elementMap, index, numAtoms, and numAtomsPerElement.
◆ readFromFile() [1/2]
| void Structure::readFromFile | ( | std::string const | fileName = "input.data" | ) |
Read configuration from file.
- Parameters
-
[in] fileName Input file name.
Reads the first configuration found in the input file.
Definition at line 97 of file Structure.cpp.
References readFromFile().
Referenced by nnp::Dataset::distributeStructures(), main(), and readFromFile().
◆ readFromFile() [2/2]
| void Structure::readFromFile | ( | std::ifstream & | file | ) |
Read configuration from file.
- Parameters
-
[in] file Input file stream (already opened).
Expects that a file with configurations is open, first keyword on first line should be begin. Reads until keyword is end.
Definition at line 113 of file Structure.cpp.
References readFromLines(), nnp::reduce(), and nnp::split().
◆ readFromLines()
| void Structure::readFromLines | ( | std::vector< std::string > const & | lines | ) |
Read configuration from lines.
- Parameters
-
[in] lines One configuration in form of a vector of strings.
Read the configuration from a vector of strings.
Definition at line 142 of file Structure.cpp.
References atoms, box, calculateInverseBox(), calculateVolume(), chargeRef, comment, elementMap, energyRef, index, isPeriodic, isTriclinic, numAtoms, numAtomsPerElement, numElements, numElementsPresent, nnp::reduce(), remap(), sampleType, nnp::split(), ST_TEST, and ST_TRAINING.
Referenced by readFromFile().
◆ calculateMaxCutoffRadiusOverall()
| void Structure::calculateMaxCutoffRadiusOverall | ( | EwaldSetup & | ewaldSetup, |
| double | rcutScreen, | ||
| double | maxCutoffRadius ) |
Calculate maximal cut-off if cut-off of screening and real part Ewald summation are also considered.
- Parameters
-
[in] ewaldSetup Settings of Ewald summation. [in] rcutScreen Cut-off for Screening of the electrostatic interaction. [in] maxCutoffRadius maximal cut-off of symmetry functions.
Definition at line 273 of file Structure.cpp.
References nnp::EwaldSetup::calculateParameters(), isPeriodic, maxCutoffRadiusOverall, numAtoms, nnp::EwaldSetup::params, nnp::EwaldParameters::rCut, and volume.
Referenced by nnp::Mode::evaluateNNP().
◆ calculateNeighborList() [1/2]
| void Structure::calculateNeighborList | ( | double | cutoffRadius, |
| bool | sortByDistance = false ) |
Calculate neighbor list for all atoms.
- Parameters
-
[in] cutoffRadius Atoms are neighbors if there distance is smaller than the cutoff radius. [in] sortByDistance Sort neighborlist from nearest to farthest neighbor.
Definition at line 287 of file Structure.cpp.
References atoms, box, calculatePbcCopies(), hasNeighborList, isPeriodic, maxCutoffRadiusOverall, nnp::Vec3D::norm(), nnp::Vec3D::norm2(), numAtoms, pbc, and sortNeighborList().
Referenced by calculateNeighborList(), nnp::Mode::evaluateNNP(), and main().
◆ calculateNeighborList() [2/2]
| void Structure::calculateNeighborList | ( | double | cutoffRadius, |
| std::vector< std::vector< double > > & | cutoffs ) |
Calculate neighbor list for all atoms and setup neighbor cut-off map.
- Parameters
-
[in] cutoffRadius Atoms are neighbors if there distance is smaller than the cutoff radius. [in] cutoffs Vector of all needed cutoffs (needed for cut-off map construction).
- Returns
- Maximum of {maxCutoffRadius, rcutScreen, rCut}.
Definition at line 400 of file Structure.cpp.
References calculateNeighborList(), and setupNeighborCutoffMap().
◆ sortNeighborList()
| void Structure::sortNeighborList | ( | ) |
Sort all neighbor lists of this structure with respect to the distance.
Definition at line 408 of file Structure.cpp.
References atoms, NeighborListIsSorted, and numAtoms.
Referenced by calculateNeighborList().
◆ setupNeighborCutoffMap()
| void Structure::setupNeighborCutoffMap | ( | std::vector< std::vector< double > > | cutoffs | ) |
Set up a neighbor cut-off map which gives the index (value) of the last needed neighbor corresponding to a specific cut-off (key).
- Parameters
-
[in] cutoffs Vector of all needed symmetry function cutoffs. Note that a local copy gets extended with maxCutoffRadiusOverall.
Definition at line 422 of file Structure.cpp.
References atoms, nnp::Atom::Neighbor::d, maxCutoffRadiusOverall, NeighborListIsSorted, and nnp::vectorContains().
Referenced by calculateNeighborList().
◆ calculatePbcCopies()
| void Structure::calculatePbcCopies | ( | double | cutoffRadius, |
| int(&) | pbc[3] ) |
Calculate required PBC copies.
- Parameters
-
[in] cutoffRadius Cutoff radius for neighbor list. [in] pbc Array for storing the result.
Called by calculateNeighborList().
Definition at line 485 of file Structure.cpp.
Referenced by calculateNeighborList().
◆ calculateInverseBox()
| void Structure::calculateInverseBox | ( | ) |
Calculate inverse box.
Simulation box looks like this:
\[h = \begin{pmatrix} \phantom{a_x} & \phantom{b_x} & \phantom{c_x} \\ \vec{\mathbf{a}} & \vec{\mathbf{b}} & \vec{\mathbf{c}} \\ \phantom{a_z} & \phantom{b_z} & \phantom{c_z} \\ \end{pmatrix} = \begin{pmatrix} a_x & b_x & c_x \\ a_y & b_y & c_y \\ a_z & b_z & c_z \\ \end{pmatrix}, \]
where \(\vec{\mathbf{a}} = \) box[0], \(\vec{\mathbf{b}} = \) box[1] and \(\vec{\mathbf{c}} = \) box[2]. Thus, indices are column first, row second:
\[h = \begin{pmatrix} \texttt{box[0][0]} & \texttt{box[1][0]} & \texttt{box[2][0]} \\ \texttt{box[0][1]} & \texttt{box[1][1]} & \texttt{box[2][1]} \\ \texttt{box[0][2]} & \texttt{box[1][2]} & \texttt{box[2][2]} \\ \end{pmatrix}. \]
The inverse box matrix (same scheme as above but with invbox) can be used to calculate fractional coordinates:
\[\begin{pmatrix} f_0 \\ f_1 \\ f_2 \end{pmatrix} = h^{-1} \; \vec{\mathbf{r}}. \]
Definition at line 518 of file Structure.cpp.
Referenced by readFromLines().
◆ canMinimumImageConventionBeApplied()
| bool Structure::canMinimumImageConventionBeApplied | ( | double | cutoffRadius | ) |
Check if cut-off radius is small enough to apply minimum image convention.
- Parameters
-
[in] cutoffRadius cut-off radius for which condition should be checked.
Definition at line 547 of file Structure.cpp.
References box.
◆ applyMinimumImageConvention()
Calculate distance between two atoms in the minimum image convention.
- Parameters
-
[in] dr Distance vector between two atoms of the same box.
Definition at line 566 of file Structure.cpp.
◆ calculateVolume()
| void Structure::calculateVolume | ( | ) |
Calculate volume from box vectors.
Definition at line 580 of file Structure.cpp.
Referenced by readFromLines().
◆ calculateElectrostaticEnergy()
| double Structure::calculateElectrostaticEnergy | ( | EwaldSetup & | ewaldSetup, |
| Eigen::VectorXd | hardness, | ||
| Eigen::MatrixXd | gammaSqrt2, | ||
| Eigen::VectorXd | sigmaSqrtPi, | ||
| ScreeningFunction const & | fs, | ||
| double const | fourPiEps, | ||
| ErfcBuf & | erfcBuf ) |
Compute electrostatic energy with global charge equilibration.
- Parameters
-
[in] ewaldSetup Settings of Ewald summation. [in] hardness Vector containing the hardness of all elements. [in] gammaSqrt2 Matrix combining gamma with prefactor. \( \text{gammaSqrt2}_{ij} = \sqrt{2} \gamma_{ij} = \sqrt{2} \sqrt{(\sigma_i^2 + \sigma_j^2)} \) [in] sigmaSqrtPi Vector combining sigma with prefactor, \( \text{sigmaSqrtPi}_i = \sqrt{\pi} \sigma_i \) [in] fs Screening function. [in] fourPiEps \( \text{fourPiEps} = 4 \pi \varepsilon_0 \). Value depends on unit system (e.g. normalization). [in] erfcBuf helper object to avoid repeated calculation of erfc().
Definition at line 587 of file Structure.cpp.
References A, atoms, box, calculateScreeningEnergy(), chargeRef, nnp::Atom::chi, nnp::Atom::element, energyElec, nnp::EwaldParameters::eta, nnp::ErfcBuf::getf(), nnp::Atom::getStoredMinNumNeighbors(), hasAMatrix, hasCharges, isPeriodic, nnp::KspaceGrid::kvectors, lambda, NeighborListIsSorted, nnp::Atom::neighbors, numAtoms, nnp::EwaldSetup::params, nnp::Atom::r, nnp::EwaldParameters::rCut, nnp::ErfcBuf::reset(), and nnp::KspaceGrid::setup().
Referenced by nnp::Mode::chargeEquilibration().
◆ calculateScreeningEnergy()
| double Structure::calculateScreeningEnergy | ( | Eigen::MatrixXd | gammaSqrt2, |
| Eigen::VectorXd | sigmaSqrtPi, | ||
| ScreeningFunction const & | fs, | ||
| double const | fourPiEps ) |
Calculate screening energy which needs to be added (!) to the electrostatic energy in order to remove contributions in the short range domain.
- Parameters
-
[in] gammaSqrt2 Matrix combining gamma with prefactor. \( \text{gammaSqrt2}_{ij} = \sqrt{2} \gamma_{ij} = \sqrt{2} \sqrt{(\sigma_i^2 + \sigma_j^2)} \) [in] sigmaSqrtPi Vector combining sigma with prefactor, \( \text{sigmaSqrtPi}_i = \sqrt{\pi} \sigma_i \) [in] fs Screening function. [in] fourPiEps \( \text{fourPiEps} = 4 \pi \varepsilon_0 \). Value depends on unit system (e.g. normalization).
Definition at line 743 of file Structure.cpp.
References A, atoms, nnp::Atom::charge, nnp::Atom::element, nnp::ScreeningFunction::f(), nnp::ScreeningFunction::getOuter(), nnp::Atom::getStoredMinNumNeighbors(), isPeriodic, nnp::Atom::neighbors, numAtoms, and nnp::Atom::r.
Referenced by calculateElectrostaticEnergy().
◆ calculateDAdrQ()
| void Structure::calculateDAdrQ | ( | EwaldSetup & | ewaldSetup, |
| Eigen::MatrixXd | gammaSqrt2, | ||
| double const | fourPiEps, | ||
| ErfcBuf & | erfcBuf ) |
Calculates derivative of A-matrix with respect to the atoms positions and contract it with Q.
- Parameters
-
[in] ewaldSetup Settings of Ewald summation. [in] gammaSqrt2 Matrix combining gamma with prefactor. \( \text{gammaSqrt2}_{ij} = \sqrt{2} \gamma_{ij} = \sqrt{2} \sqrt{(\sigma_i^2 + \sigma_j^2)} \) [in] fourPiEps \( \text{fourPiEps} = 4 \pi \varepsilon_0 \). Value depends on unit system (e.g. normalization). [in] erfcBuf helper object to avoid repeated calculation of erfc().
Definition at line 841 of file Structure.cpp.
References atoms, box, nnp::Atom::charge, nnp::Atom::dAdrQ, nnp::Atom::element, nnp::EwaldParameters::eta, nnp::ErfcBuf::getf(), nnp::Atom::getStoredMinNumNeighbors(), isPeriodic, nnp::KspaceGrid::kvectors, nnp::Atom::neighbors, numAtoms, nnp::EwaldSetup::params, nnp::Atom::r, nnp::EwaldParameters::rCut, and nnp::KspaceGrid::setup().
Referenced by nnp::Mode::chargeEquilibration().
◆ calculateDQdChi()
| void Structure::calculateDQdChi | ( | std::vector< Eigen::VectorXd > & | dQdChi | ) |
Calculates derivative of the charges with respect to electronegativities.
- Parameters
-
[in] dQdChi vector to store the result. dQdChi[i](j) represents the derivative for the i-th electronegativity and the j-th charge.
Definition at line 1001 of file Structure.cpp.
Referenced by nnp::Training::update().
◆ calculateDQdJ()
| void Structure::calculateDQdJ | ( | std::vector< Eigen::VectorXd > & | dQdJ | ) |
Calculates derivative of the charges with respect to atomic hardness.
- Parameters
-
[in] dQdJ vector to store the result. dQdJ[i](j) represents the derivative for the i-th hardness and the j-th charge.
Definition at line 1016 of file Structure.cpp.
References A, atoms, nnp::Atom::charge, nnp::Atom::element, numAtoms, and numElements.
Referenced by nnp::Training::update().

◆ calculateDQdr()
| void Structure::calculateDQdr | ( | std::vector< size_t > const & | atomsIndices, |
| std::vector< size_t > const & | compIndices, | ||
| double const | maxCutoffRadius, | ||
| std::vector< Element > const & | elements ) |
Calculates derivative of the charges with respect to the atom's position.
- Parameters
-
[in] atomIndices Vector containing indices of atoms for which the derivative should be calculated. [in] compIndices Vector containing indices of vector components for which the derivative should be calculated. [in] maxCutoffRadius Max. cutoff radius of symmetry functions. [in] elements Vector containing all elements (needed for symmetry function table).
Definition at line 1035 of file Structure.cpp.
References A, atoms, nnp::Atom::calculateDChidr(), nnp::Atom::dAdrQ, nnp::Atom::dQdr, nnp::Atom::element, and numAtoms.
Referenced by nnp::Training::update().


◆ calculateElectrostaticEnergyDerivatives()
| void Structure::calculateElectrostaticEnergyDerivatives | ( | Eigen::VectorXd | hardness, |
| Eigen::MatrixXd | gammaSqrt2, | ||
| Eigen::VectorXd | sigmaSqrtPi, | ||
| ScreeningFunction const & | fs, | ||
| double const | fourPiEps ) |
Calculates partial derivatives of electrostatic Energy with respect to atom's coordinates and charges.
- Parameters
-
[in] hardness Vector containing the hardness of all elements. [in] gammaSqrt2 Matrix combining gamma with prefactor. \( \text{gammaSqrt2}_{ij} = \sqrt{2} \gamma_{ij} = \sqrt{2} \sqrt{(\sigma_i^2 + \sigma_j^2)} \) [in] sigmaSqrtPi Vector combining sigma with prefactor, \( \text{sigmaSqrtPi}_i = \sqrt{\pi} \sigma_i \) [in] fs Screening function. [in] fourPiEps \( \text{fourPiEps} = 4 \pi \varepsilon_0 \). Value depends on unit system (e.g. normalization).
Definition at line 1079 of file Structure.cpp.
References A, atoms, nnp::Atom::charge, nnp::Atom::dAdrQ, nnp::Atom::dEelecdQ, nnp::ScreeningFunction::df(), nnp::Atom::element, nnp::ScreeningFunction::f(), nnp::ScreeningFunction::getOuter(), isPeriodic, nnp::Atom::neighbors, numAtoms, nnp::Atom::pEelecpr, and nnp::Atom::r.
Referenced by nnp::Mode::chargeEquilibration().
◆ calculateForceLambdaTotal()
| VectorXd const Structure::calculateForceLambdaTotal | ( | ) | const |
Calculate lambda_total vector which is needed for the total force calculation in 4G NN.
Definition at line 1184 of file Structure.cpp.
References A, atoms, nnp::Atom::dEdG, nnp::Atom::dEelecdQ, and numAtoms.
Referenced by nnp::Mode::calculateForces(), and nnp::InterfaceLammps::getForcesDevelop().
◆ calculateForceLambdaElec()
| VectorXd const Structure::calculateForceLambdaElec | ( | ) | const |
Calculate lambda_elec vector which is needed for the electrostatic force calculation in 4G NN.
Definition at line 1197 of file Structure.cpp.
References A, atoms, nnp::Atom::dEelecdQ, and numAtoms.
Referenced by nnp::Mode::calculateForces().
◆ remap() [1/2]
| void Structure::remap | ( | ) |
Translate all atoms back into box if outside.
Definition at line 1210 of file Structure.cpp.
References atoms, and remap().
Referenced by nnp::Dataset::prepareNumericForces(), readFromLines(), and remap().
◆ remap() [2/2]
| void Structure::remap | ( | Atom & | atom | ) |
Translate atom back into box if outside.
- Parameters
-
[in,out] atom Atom to be remapped.
Definition at line 1219 of file Structure.cpp.
References box, invbox, and nnp::Atom::r.
◆ toNormalizedUnits()
| void Structure::toNormalizedUnits | ( | double | meanEnergy, |
| double | convEnergy, | ||
| double | convLength, | ||
| double | convCharge ) |
Normalize structure, shift energy and change energy, length and charge unit.
- Parameters
-
[in] meanEnergy Mean energy per atom (in old units). [in] convEnergy Multiplicative energy unit conversion factor. [in] convLength Multiplicative length unit conversion factor. [in] convCharge Multiplicative charge unit conversion factor.
Definition at line 1245 of file Structure.cpp.
References atoms, box, charge, chargeRef, energy, energyRef, invbox, isPeriodic, numAtoms, and volume.
Referenced by nnp::Mode::convertToNormalizedUnits().
◆ toPhysicalUnits()
| void Structure::toPhysicalUnits | ( | double | meanEnergy, |
| double | convEnergy, | ||
| double | convLength, | ||
| double | convCharge ) |
Switch to physical units, shift energy and change energy, length and charge unit.
- Parameters
-
[in] meanEnergy Mean energy per atom (in old units). [in] convEnergy Multiplicative energy unit conversion factor. [in] convLength Multiplicative length unit conversion factor. [in] convCharge Multiplicative charge unit conversion factor.
Definition at line 1274 of file Structure.cpp.
References atoms, box, charge, chargeRef, energy, energyRef, invbox, isPeriodic, numAtoms, and volume.
Referenced by nnp::Mode::convertToPhysicalUnits().
◆ getMaxNumNeighbors()
| size_t Structure::getMaxNumNeighbors | ( | ) | const |
Find maximum number of neighbors.
- Returns
- Maximum numbor of neighbors of all atoms in this structure.
Definition at line 472 of file Structure.cpp.
References atoms.
◆ freeAtoms()
| void Structure::freeAtoms | ( | bool | all, |
| double const | maxCutoffRadius = 0.0 ) |
Free symmetry function memory for all atoms, see free() in Atom class.
- Parameters
-
[in] all See description in Atom. [in] maxCutoffRadius Maximum cutoff radius of symmetry functions.
Definition at line 1303 of file Structure.cpp.
References atoms, hasSymmetryFunctionDerivatives, and hasSymmetryFunctions.
Referenced by nnp::Training::update().

◆ reset()
| void Structure::reset | ( | ) |
Reset everything but elementMap.
Definition at line 1315 of file Structure.cpp.
References atoms, box, charge, chargeRef, comment, elementMap, energy, energyRef, hasNeighborList, hasSymmetryFunctionDerivatives, hasSymmetryFunctions, index, invbox, isPeriodic, isTriclinic, numAtoms, numAtomsPerElement, numElements, numElementsPresent, pbc, sampleType, ST_UNKNOWN, and volume.
Referenced by main().
◆ clearNeighborList()
| void Structure::clearNeighborList | ( | ) |
Clear neighbor list of all atoms.
Definition at line 1352 of file Structure.cpp.
References atoms, nnp::Atom::clearNeighborList(), hasNeighborList, hasSymmetryFunctionDerivatives, hasSymmetryFunctions, numAtoms, numElements, and nnp::Atom::numNeighborsPerElement.
◆ clearElectrostatics()
| void Structure::clearElectrostatics | ( | bool | clearDQdr = false | ) |
Clear A-matrix, dAdrQ and optionally dQdr.
- Parameters
-
[in] clearDqdr Specify if dQdr should also be cleared.
Definition at line 1368 of file Structure.cpp.
References A, atoms, and hasAMatrix.
Referenced by nnp::Training::update().

◆ updateError()
| void Structure::updateError | ( | std::string const & | property, |
| std::map< std::string, double > & | error, | ||
| std::size_t & | count ) const |
Update property error metrics with this structure.
- Parameters
-
[in] property One of "energy", "force" or "charge". [in,out] error Input error metric map to be updated. [in,out] count Input counter to be updated.
The "energy" error metric map stores temporary sums for the following metrics:
key "RMSEpa": RMSE of energy per atom key "RMSE" : RMSE of energy key "MAEpa" : MAE of energy per atom key "MAE" : MAE of energy
The "force" error metric map stores temporary sums for the following metrics:
key "RMSE" : RMSE of forces key "MAE" : MAE of forces
The "charge" error metric map stores temporary sums for the following metrics:
key "RMSE" : RMSE of charges key "MAE" : MAE of charges
Definition at line 1382 of file Structure.cpp.
◆ getEnergyLine()
| string Structure::getEnergyLine | ( | ) | const |
Get reference and NN energy.
Definition at line 1412 of file Structure.cpp.
References energy, energyRef, index, numAtoms, and nnp::strpr().

◆ getForcesLines()
| vector< string > Structure::getForcesLines | ( | ) | const |
Get reference and NN forces for all atoms.
- Returns
- Vector of strings with force comparison.
Definition at line 1420 of file Structure.cpp.
References atoms.
◆ getChargesLines()
| vector< string > Structure::getChargesLines | ( | ) | const |
Get reference and NN charges for all atoms.
- Returns
- Vector of strings with charge comparison.
Definition at line 1433 of file Structure.cpp.
References atoms.
◆ writeToFile() [1/2]
| void Structure::writeToFile | ( | std::string const | fileName = "output.data", |
| bool const | ref = true, | ||
| bool const | append = false ) const |
Write configuration to file.
- Parameters
-
[in,out] fileName Ouptut file name. [in] ref If true, write reference energy and forces, if false, write NNP results instead. [in] append If true, append to existing file.
Definition at line 1445 of file Structure.cpp.
References writeToFile().
Referenced by main(), and writeToFile().

◆ writeToFile() [2/2]
| void Structure::writeToFile | ( | std::ofstream *const & | file, |
| bool const | ref = true ) const |
Write configuration to file.
- Parameters
-
[in,out] file Ouptut file. [in] ref If true, write reference energy and forces, if false, write NNP results instead.
Definition at line 1470 of file Structure.cpp.
References atoms, box, charge, chargeRef, comment, elementMap, energy, energyRef, isPeriodic, and nnp::strpr().
◆ writeToFileXyz()
| void Structure::writeToFileXyz | ( | std::ofstream *const & | file | ) | const |
Write configuration to xyz file.
- Parameters
-
[in,out] file xyz output file.
Definition at line 1529 of file Structure.cpp.
References atoms, box, elementMap, isPeriodic, numAtoms, and nnp::strpr().
Referenced by main().


◆ writeToFilePoscar() [1/2]
| void Structure::writeToFilePoscar | ( | std::ofstream *const & | file | ) | const |
Write configuration to POSCAR file.
- Parameters
-
[in,out] file POSCAR output file.
- Warning
- Elements in POTCAR file must be ordered according to periodic table.
Definition at line 1564 of file Structure.cpp.
References elementMap, and writeToFilePoscar().
Referenced by main(), and writeToFilePoscar().

◆ writeToFilePoscar() [2/2]
| void Structure::writeToFilePoscar | ( | std::ofstream *const & | file, |
| std::string const | elements ) const |
Write configuration to POSCAR file.
- Parameters
-
[in,out] file POSCAR output file. [in,out] elements User-defined order of elements, e.g. "Zn O Cu".
Definition at line 1571 of file Structure.cpp.
References atoms, box, comment, elementMap, isPeriodic, numAtomsPerElement, nnp::split(), and nnp::strpr().
◆ info()
| vector< string > Structure::info | ( | ) | const |
Get structure information as a vector of strings.
- Returns
- Lines with structure information.
Definition at line 1637 of file Structure.cpp.
References atoms, box, charge, chargeRef, comment, elementMap, energy, energyRef, hasNeighborList, hasSymmetryFunctionDerivatives, hasSymmetryFunctions, index, invbox, isPeriodic, isTriclinic, numAtoms, numAtomsPerElement, numElements, numElementsPresent, pbc, sampleType, nnp::strpr(), and volume.
Referenced by main().


Member Data Documentation
◆ elementMap
| ElementMap nnp::Structure::elementMap |
Copy of element map provided as constructor argument.
Definition at line 59 of file Structure.h.
Referenced by addAtom(), info(), readFromLines(), reset(), setElementMap(), writeToFile(), writeToFilePoscar(), writeToFilePoscar(), and writeToFileXyz().
◆ isPeriodic
| bool nnp::Structure::isPeriodic |
If periodic boundary conditions apply.
Definition at line 61 of file Structure.h.
Referenced by calculateDAdrQ(), calculateElectrostaticEnergy(), calculateElectrostaticEnergyDerivatives(), calculateMaxCutoffRadiusOverall(), calculateNeighborList(), calculateScreeningEnergy(), info(), main(), nnp::Dataset::prepareNumericForces(), readFromLines(), nnp::Dataset::recvStructure(), reset(), nnp::Dataset::sendStructure(), Structure(), toNormalizedUnits(), toPhysicalUnits(), writeToFile(), writeToFilePoscar(), and writeToFileXyz().
◆ isTriclinic
| bool nnp::Structure::isTriclinic |
If the simulation box is triclinic.
Definition at line 63 of file Structure.h.
Referenced by info(), readFromLines(), nnp::Dataset::recvStructure(), reset(), nnp::Dataset::sendStructure(), and Structure().
◆ hasNeighborList
| bool nnp::Structure::hasNeighborList |
If the neighbor list has been calculated.
Definition at line 65 of file Structure.h.
Referenced by calculateNeighborList(), clearNeighborList(), info(), nnp::Dataset::recvStructure(), reset(), nnp::Dataset::sendStructure(), and Structure().
◆ NeighborListIsSorted
| bool nnp::Structure::NeighborListIsSorted |
If the neighbor list has been sorted by distance.
Definition at line 67 of file Structure.h.
Referenced by calculateElectrostaticEnergy(), setupNeighborCutoffMap(), sortNeighborList(), and Structure().
◆ hasSymmetryFunctions
| bool nnp::Structure::hasSymmetryFunctions |
If symmetry function values are saved for each atom.
Definition at line 69 of file Structure.h.
Referenced by nnp::Mode::calculateSymmetryFunctionGroups(), nnp::Mode::calculateSymmetryFunctions(), clearNeighborList(), freeAtoms(), info(), nnp::Dataset::recvStructure(), reset(), nnp::Dataset::sendStructure(), and Structure().
◆ hasSymmetryFunctionDerivatives
| bool nnp::Structure::hasSymmetryFunctionDerivatives |
If symmetry function derivatives are saved for each atom.
Definition at line 71 of file Structure.h.
Referenced by nnp::Mode::calculateSymmetryFunctionGroups(), nnp::Mode::calculateSymmetryFunctions(), clearNeighborList(), freeAtoms(), info(), nnp::Dataset::recvStructure(), reset(), nnp::Dataset::sendStructure(), and Structure().
◆ index
| std::size_t nnp::Structure::index |
Index number of this structure.
Definition at line 73 of file Structure.h.
Referenced by addAtom(), nnp::Dataset::distributeStructures(), getEnergyLine(), info(), main(), readFromLines(), nnp::Dataset::recvStructure(), reset(), nnp::Training::selectSets(), nnp::Dataset::sendStructure(), and Structure().
◆ numAtoms
| std::size_t nnp::Structure::numAtoms |
Total number of atoms present in this structure.
Definition at line 75 of file Structure.h.
Referenced by addAtom(), nnp::Training::calculateChargeErrorVec(), calculateDAdrQ(), calculateDQdChi(), calculateDQdJ(), calculateDQdr(), calculateElectrostaticEnergy(), calculateElectrostaticEnergyDerivatives(), calculateForceLambdaElec(), calculateForceLambdaTotal(), nnp::Mode::calculateForces(), calculateMaxCutoffRadiusOverall(), calculateNeighborList(), calculateScreeningEnergy(), clearNeighborList(), nnp::Training::dPdc(), nnp::Training::dPdcN(), nnp::InterfaceLammps::getCharges(), getEnergyLine(), nnp::InterfaceLammps::getForcesDevelop(), info(), main(), nnp::Mode::normalizedEnergy(), nnp::Mode::physicalEnergy(), nnp::Dataset::prepareNumericForces(), readFromLines(), nnp::Dataset::recvStructure(), reset(), nnp::Training::selectSets(), nnp::Dataset::sendStructure(), sortNeighborList(), nnp::Training::sortUpdateCandidates(), Structure(), toNormalizedUnits(), toPhysicalUnits(), nnp::Training::update(), updateError(), and writeToFileXyz().
◆ numElements
| std::size_t nnp::Structure::numElements |
Global number of elements (all structures).
Definition at line 77 of file Structure.h.
Referenced by calculateDQdJ(), clearNeighborList(), info(), main(), readFromLines(), nnp::Dataset::recvStructure(), reset(), nnp::Dataset::sendStructure(), setElementMap(), and Structure().
◆ numElementsPresent
| std::size_t nnp::Structure::numElementsPresent |
Number of elements present in this structure.
Definition at line 79 of file Structure.h.
Referenced by info(), readFromLines(), nnp::Dataset::recvStructure(), reset(), nnp::Dataset::sendStructure(), and Structure().
◆ pbc
| int nnp::Structure::pbc[3] |
Number of PBC images necessary in each direction for max cut-off.
Definition at line 81 of file Structure.h.
Referenced by calculateNeighborList(), calculatePbcCopies(), info(), nnp::Dataset::recvStructure(), reset(), nnp::Dataset::sendStructure(), and Structure().
◆ energy
| double nnp::Structure::energy |
Potential energy determined by neural network.
Definition at line 83 of file Structure.h.
Referenced by nnp::Mode::addEnergyOffset(), nnp::Mode::calculateEnergy(), nnp::Training::dPdcN(), getEnergyLine(), nnp::Mode::getEnergyWithOffset(), info(), main(), nnp::Mode::normalizedEnergy(), nnp::Mode::physicalEnergy(), nnp::Dataset::recvStructure(), nnp::Mode::removeEnergyOffset(), reset(), nnp::Dataset::sendStructure(), nnp::Training::sortUpdateCandidates(), Structure(), toNormalizedUnits(), toPhysicalUnits(), nnp::Training::update(), updateError(), and writeToFile().
◆ energyRef
| double nnp::Structure::energyRef |
Reference potential energy.
Definition at line 85 of file Structure.h.
Referenced by nnp::Mode::addEnergyOffset(), getEnergyLine(), nnp::Mode::getEnergyWithOffset(), info(), main(), nnp::Mode::normalizedEnergy(), nnp::Mode::physicalEnergy(), readFromLines(), nnp::Dataset::recvStructure(), nnp::Mode::removeEnergyOffset(), reset(), nnp::Dataset::sendStructure(), nnp::Training::sortUpdateCandidates(), Structure(), toNormalizedUnits(), toPhysicalUnits(), nnp::Training::update(), updateError(), and writeToFile().
◆ energyShort
| double nnp::Structure::energyShort |
Short-range part of the potential energy predicted by NNP.
Definition at line 88 of file Structure.h.
Referenced by nnp::Mode::calculateEnergy(), and Structure().
◆ energyElec
| double nnp::Structure::energyElec |
Electrostatics part of the potential energy predicted by NNP.
Definition at line 91 of file Structure.h.
Referenced by calculateElectrostaticEnergy(), nnp::Mode::calculateEnergy(), main(), and Structure().
◆ hasCharges
| bool nnp::Structure::hasCharges |
If all charges of this structure have been calculated (and stay the same, e.g.
for stage 2).
Definition at line 94 of file Structure.h.
Referenced by calculateElectrostaticEnergy(), Structure(), and nnp::Training::update().
◆ charge
| double nnp::Structure::charge |
Charge determined by neural network potential.
Definition at line 96 of file Structure.h.
Referenced by nnp::Mode::calculateCharge(), info(), main(), nnp::Dataset::recvStructure(), reset(), nnp::Dataset::sendStructure(), Structure(), toNormalizedUnits(), toPhysicalUnits(), and writeToFile().
◆ chargeRef
| double nnp::Structure::chargeRef |
Reference charge.
Definition at line 98 of file Structure.h.
Referenced by calculateElectrostaticEnergy(), info(), main(), readFromLines(), nnp::Dataset::recvStructure(), reset(), nnp::Dataset::sendStructure(), Structure(), toNormalizedUnits(), toPhysicalUnits(), and writeToFile().
◆ volume
| double nnp::Structure::volume |
Simulation box volume.
Definition at line 100 of file Structure.h.
Referenced by calculateMaxCutoffRadiusOverall(), calculateVolume(), info(), main(), nnp::Dataset::recvStructure(), reset(), nnp::Dataset::sendStructure(), Structure(), toNormalizedUnits(), and toPhysicalUnits().
◆ maxCutoffRadiusOverall
| double nnp::Structure::maxCutoffRadiusOverall |
Maximum cut-off radius with respect to symmetry functions, screening function and Ewald summation.
Definition at line 103 of file Structure.h.
Referenced by calculateMaxCutoffRadiusOverall(), calculateNeighborList(), setupNeighborCutoffMap(), and Structure().
◆ lambda
| double nnp::Structure::lambda |
Lagrange multiplier used for charge equilibration.
Definition at line 106 of file Structure.h.
Referenced by calculateElectrostaticEnergy().
◆ sampleType
| SampleType nnp::Structure::sampleType |
Sample type (training or test set).
Definition at line 108 of file Structure.h.
Referenced by info(), readFromLines(), nnp::Dataset::recvStructure(), reset(), nnp::Training::selectSets(), nnp::Dataset::sendStructure(), and Structure().
◆ comment
| std::string nnp::Structure::comment |
Structure comment.
Definition at line 110 of file Structure.h.
Referenced by nnp::Dataset::calculateBufferSize(), info(), main(), nnp::Dataset::prepareNumericForces(), readFromLines(), nnp::Dataset::recvStructure(), reset(), nnp::Dataset::sendStructure(), Structure(), writeToFile(), and writeToFilePoscar().
◆ box
| Vec3D nnp::Structure::box[3] |
Simulation box vectors.
Definition at line 112 of file Structure.h.
Referenced by applyMinimumImageConvention(), calculateDAdrQ(), calculateElectrostaticEnergy(), calculateInverseBox(), calculateNeighborList(), calculatePbcCopies(), calculateVolume(), canMinimumImageConventionBeApplied(), info(), main(), readFromLines(), nnp::Dataset::recvStructure(), remap(), reset(), nnp::Dataset::sendStructure(), Structure(), toNormalizedUnits(), toPhysicalUnits(), writeToFile(), writeToFilePoscar(), and writeToFileXyz().
◆ invbox
| Vec3D nnp::Structure::invbox[3] |
Inverse simulation box vectors.
Definition at line 114 of file Structure.h.
Referenced by applyMinimumImageConvention(), calculateInverseBox(), info(), nnp::Dataset::recvStructure(), remap(), reset(), nnp::Dataset::sendStructure(), Structure(), toNormalizedUnits(), and toPhysicalUnits().
◆ A
| Eigen::MatrixXd nnp::Structure::A |
Global charge equilibration matrix A'.
Definition at line 116 of file Structure.h.
Referenced by calculateDQdChi(), calculateDQdJ(), calculateDQdr(), calculateElectrostaticEnergy(), calculateElectrostaticEnergyDerivatives(), calculateForceLambdaElec(), calculateForceLambdaTotal(), calculateScreeningEnergy(), and clearElectrostatics().
◆ hasAMatrix
| bool nnp::Structure::hasAMatrix |
If A matrix of this structure is currently stored.
Definition at line 118 of file Structure.h.
Referenced by calculateElectrostaticEnergy(), clearElectrostatics(), Structure(), and nnp::Training::update().
◆ numAtomsPerElement
| std::vector<std::size_t> nnp::Structure::numAtomsPerElement |
Number of atoms of each element in this structure.
Definition at line 120 of file Structure.h.
Referenced by addAtom(), nnp::Mode::addEnergyOffset(), nnp::Dataset::calculateBufferSize(), nnp::Mode::getEnergyOffset(), nnp::Mode::getEnergyWithOffset(), info(), main(), readFromLines(), nnp::Dataset::recvStructure(), nnp::Mode::removeEnergyOffset(), reset(), nnp::Training::selectSets(), nnp::Dataset::sendStructure(), setElementMap(), nnp::Training::update(), and writeToFilePoscar().
◆ atoms
| std::vector<Atom> nnp::Structure::atoms |
Vector of all atoms in this structure.
Definition at line 122 of file Structure.h.
Referenced by addAtom(), nnp::Mode::calculateAtomicNeuralNetworks(), nnp::Dataset::calculateBufferSize(), nnp::Mode::calculateCharge(), nnp::Training::calculateChargeErrorVec(), calculateDAdrQ(), calculateDQdJ(), calculateDQdr(), calculateElectrostaticEnergy(), calculateElectrostaticEnergyDerivatives(), nnp::Mode::calculateEnergy(), calculateForceLambdaElec(), calculateForceLambdaTotal(), nnp::Mode::calculateForces(), calculateNeighborList(), calculateScreeningEnergy(), nnp::Mode::calculateSymmetryFunctionGroups(), nnp::Mode::calculateSymmetryFunctions(), nnp::Training::calculateWeightDerivatives(), nnp::Training::calculateWeightDerivatives(), clearElectrostatics(), clearNeighborList(), nnp::Training::dPdc(), nnp::Training::dPdcN(), nnp::Mode::evaluateNNP(), freeAtoms(), nnp::InterfaceLammps::getCharges(), getChargesLines(), nnp::InterfaceLammps::getForcesDevelop(), getForcesLines(), getMaxNumNeighbors(), info(), main(), nnp::Dataset::prepareNumericForces(), readFromLines(), nnp::Dataset::recvStructure(), remap(), reset(), nnp::Training::selectSets(), nnp::Dataset::sendStructure(), setupNeighborCutoffMap(), sortNeighborList(), nnp::Training::sortUpdateCandidates(), toNormalizedUnits(), toPhysicalUnits(), nnp::Training::update(), updateError(), writeToFile(), writeToFilePoscar(), and writeToFileXyz().
The documentation for this struct was generated from the following files:
- /home/runner/work/n2p2/n2p2/src/libnnp/Structure.h
- /home/runner/work/n2p2/n2p2/src/libnnp/Structure.cpp